Le calcul des propriétés électroniques des matériaux organiques est une clef essentielle pour la compréhension et la maîtrise des mécanismes physiques à l’œuvre dans les dispositifs de type photovoltaïque (PV) organique ou les OLED (diode électro-luminescente organique). Que ce soit pour l’étude des propriétés optiques de ces matériaux, ou plus indirectement pour celle des propriétés de transport,
via les mécanismes de dopage notamment, les chercheurs de l’Irig développent depuis plusieurs années des méthodologies basées sur les théories de perturbation à N corps, et plus précisément la théorie GW permettant d’accéder
ab initio (
i.e. à partir de principes premiers) aux grandeurs d’intérêt pour des systèmes physiques d’intérêt expérimental.
La méthode de calcul des structures électroniques la moins coûteuse numériquement et la plus utilisée (la DFT,
Density Functional Theory), s’applique uniquement à des systèmes qui se trouvent dans leur état fondamental. La méthode GW permet quant à elle de calculer les excitations des molécules comme les spectres d’absorption, de photo-émission ou la fluorescence. Mais le calcul des excitations d’une molécule de 100 atomes par la méthode GW, bien que possible, requiert l’utilisation de super-ordinateurs.
Fruit d’une collaboration avec l’institut Néel, un ensemble d’innovations théoriques nous a permis de réduire significativement la complexité calculatoire de la méthode GW. Couplés à d’autres méthodes mises en place, nos nouveaux développements ouvrent pour la première fois la voie à la simulation de systèmes de très grandes tailles (de l'ordre du millier d’atomes) dans un environnement électrostatique complexe.
Ces développements, implémentés dans le code de calcul massivement parallèle beDeft, font actuellement l’objet d’un grand challenge sur l’extension AMD Rome du supercalculateur Irène du Très grand centre de calcul du CEA (TGCC) à Bruyères-le-Châtel, avec pour finalité la démonstration du premier calcul GW tout électron sur un système d’un millier d’atomes.
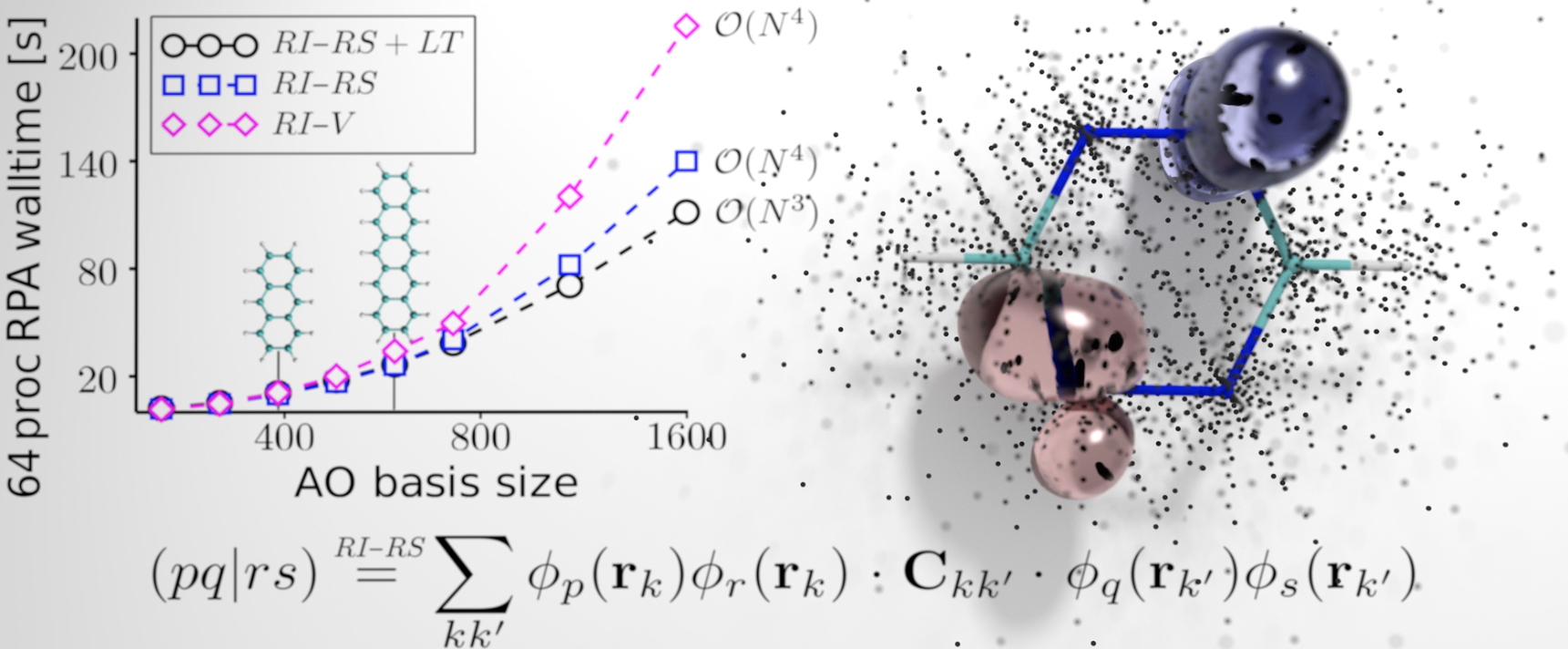
Coût numérique de différentes méthodes GW pour le calcul des interactions coulombiennes en fonction du nombre d'orbitales atomiques. Illustration de la méthode GW en espace réel avec le nuage de points considérés pour la molécule de benzène.
Ce travail s’appuie ainsi sur deux innovations fondamentales : la formulation d’une technique de résolution de l’identité séparable, basée sur une approche en espace réel (illustration) d’une part, et d’autre part l’application des techniques d’analyse complexe dites de continuation analytique à la description du potentiel coulombien écranté (W), ingrédient central de la méthodologie GW.